
To truly understand the problems with development of drugs specific to childhood cancer, and rare disease in general, examine the FDA’s Therapeutic Development Pipeline below. The pipeline represents the normal development of an average drug. The average successful drug will spend 14 years in the pipeline from Drug Discovery to Pre-clinical, through Clinical Trials and the FDA Review process before it is approved and available for use in the Clinic.
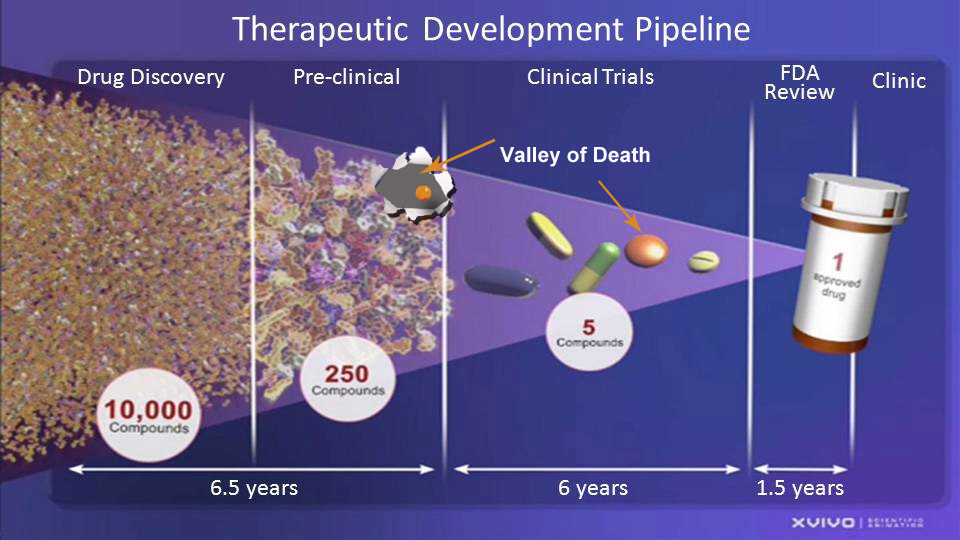
The average drug spends 6 and 1/2 years in Drug Discovery and Preclinical testing which is mostly funded by the government’s National Institute of Health (NIH) in the form of grants. Private foundations also provide a large amount of funding trying to produce an effective child specific cancer drug.
 Some call the next area “The Valley of Death” for child specific cancer drugs because this step requires funding from a private sponsor which is generally defined as a venture capitalist, manufacturer, corporation or a private institution but not the government. If a sponsor determines there is an opportunity to make a profit, an investigational new drug application (IND) is made to the FDA. It outlines what the sponsor of a new drug proposes for human testing. The FDA then approves the drug for clinical trials. Since the
Some call the next area “The Valley of Death” for child specific cancer drugs because this step requires funding from a private sponsor which is generally defined as a venture capitalist, manufacturer, corporation or a private institution but not the government. If a sponsor determines there is an opportunity to make a profit, an investigational new drug application (IND) is made to the FDA. It outlines what the sponsor of a new drug proposes for human testing. The FDA then approves the drug for clinical trials. Since the  childhood cancer area is considered to be a very small market, it is usually considered to be unattractive to sponsors because they can not justify the investment risk for the amount of return they anticipate. If a sponsor cannot be located, the research dies right here, and does not move forward to clinical trials.
childhood cancer area is considered to be a very small market, it is usually considered to be unattractive to sponsors because they can not justify the investment risk for the amount of return they anticipate. If a sponsor cannot be located, the research dies right here, and does not move forward to clinical trials.
Clinical trials average another 6 years in the overall process. The first phase of the trials involve a small population of people and determines if the drug is safe for use by humans. Phase I does not determine the effectiveness of the drug. After it is determined that the drug is safe for use, the next two phases concentrate on the drugs effectiveness, proper dosage and side effects related to use of the drug. Phase II can involve a few dozen to 300 people while Phase III may involve as many as 300 to 3,000 people for the average adult drug.
Because children’s cancer cases are in small numbers relative to adults, if there are not enough people to complete clinical trials by FDA standards, studies may be done by Children’s Oncology Group (COG) and if they see promising results and of benefit to children fighting cancer, the drug will proceed forward.
The FDA Review Process starts with a pre-NDA period, just before a new drug application (NDA) is submitted. This is a common time for the FDA and drug sponsors to meet. Next, the NDA is submitted. It is the formal step asking the FDA to consider a drug for marketing approval. The FDA has 60 days to decide whether to file the NDA so it can be reviewed. If the FDA files the NDA, a review team is assigned to evaluate the sponsor’s research on the drug’s safety and effectiveness.
The FDA also reviews information that goes on a drug’s professional labeling (information on how to use the drug) and it inspects the facilities where the drug will be manufactured as part of the approval process. FDA reviewers will approve the application or issue a complete response letter. The average amount of time for the above required FDA steps is approximately 1 1/2 years for the average drug.
Considering all the above, it takes 14 years for the average drug from beginning to end. What is even more shocking is that the failure rate exceeds 95 percent, and after adjusting for all of the failures, the cost per successful drug is estimated to be between $1.5 billion to $2 billion!

Author: Joe Baber
Editor’s Note: Nancy Goodman, Kids V Cancer, is the champion of the Creating Hope Act. It was signed into law in June 2012 and it gives companies certain incentives to develop drugs specifically for childhood cancer. She has several companies now in discussions with the FDA that may result in the creation of new drugs in the future. Three companies have already received large financial benefit from FDA fast track vouchers as a result of the act. Nancy is continuing to work with the FDA to adopt policies that maximize the potential for pediatric cancer  drug development. She is working to identify areas of potential convergence between FDA and foreign regulatory practices that can help strengthen pediatric drug development. She continues to work with researchers, pharmaceutical companies and biotech firms to enhance access to promising drugs.
drug development. She is working to identify areas of potential convergence between FDA and foreign regulatory practices that can help strengthen pediatric drug development. She continues to work with researchers, pharmaceutical companies and biotech firms to enhance access to promising drugs.
Related Articles:
Engaging the Phamaceutical Industry By Jonathan Agin.















Well wriiten! Would like to be on your mailing list.
Angie, Your will get an email from us. Please approve the email verifying that you want to get the auto updates. Once that is done, you will get all the updates as they occur. Thanks for your interest.
Pingback: Engaging the Pharmaceutical Industry – Part 1 | Four-Square Clobbers Cancer